Oral Sphere
Journal of Dental and Health Sciences
Journal of Dental and Health Sciences
Received: 2025-07-07
Accepted: 2025-09-25
Published: 2025-10-01
Pages: 244-258
Background: Irinotecan, a vital chemotherapeutic in pediatric oncology, often causes severe toxicities, including grade 3-4 neutropenia and diarrhoea. These adverse events are strongly linked to interpatient variability in the metabolism of its active metabolite, SN-38, primarily by the UDP-glucuronosyltransferase 1A1 (UGT1A1) enzyme. Polymorphisms like UGT1A1*28 and *6 impair this detoxification, increasing toxicity risk. While UGT1A1 genotyping is standard for adults, pediatric-specific guidelines are lacking due to developmental differences in enzyme expression and pharmacokinetics. This systematic review aims to synthesize evidence on UGT1A1 pharmacogenomics in pediatric cancer patients receiving irinotecan, focusing on genotype-toxicity associations, implementation challenges, and research gaps.
Methodology: Following PRISMA guidelines, we searched PubMed, Scopus, and EMBASE (January 2000 – August 2025). Included studies involved pediatric patients (<18 years) with irinotecan treatment, reporting UGT1A1 genotypes and their association with grade 3-4 neutropenia or diarrhoea. Data were narratively synthesized, and study quality assessed.
Results: Twenty-six studies (n=2,158 pediatric patients) consistently confirmed that UGT1A1*28 and *6 polymorphisms significantly increase the risk of severe neutropenia and diarrhoea (ORs typically 2.5-4.5). Genotype effects were attenuated in younger children (<5 years) due to developmental variations in UGT1A1 expression. Implementation barriers included testing cost, limited pediatric guidelines, and clinician unfamiliarity.
Conclusion: UGT1A1 polymorphisms are strong predictors of severe irinotecan toxicity in pediatric cancer patients. Pretherapeutic genotyping offers significant potential for personalized dosing and toxicity reduction. Urgent development of age-stratified guidelines and addressing implementation challenges are crucial for advancing precision medicine in pediatric oncology.
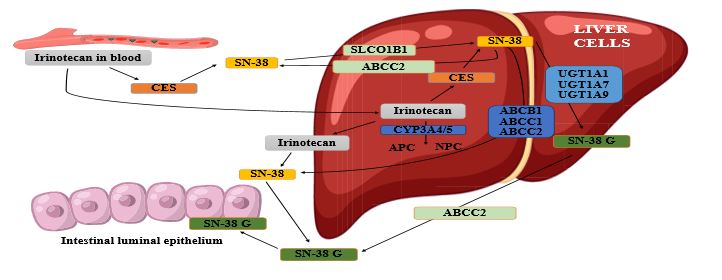
Irinotecan (CPT-11), a semi-synthetic camptothecin analog, stands as a cornerstone in combination chemotherapy for a diverse array of pediatric malignancies. Its efficacy is particularly notable in high-risk neuroblastoma, rhabdomyosarcoma, Wilms tumor, and various refractory solid tumors [1]. The drug functions as a prodrug, undergoing metabolism in the liver by carboxylesterases into its highly potent and cytotoxic metabolite, SN-38. SN-38 exerts its antineoplastic effect by inhibiting topoisomerase I, a critical enzyme involved in DNA replication and repair, thereby disrupting cancer cell proliferation [2].
Despite its undisputed therapeutic benefits, the clinical application of irinotecan is significantly challenged by pronounced interpatient variability in its pharmacokinetics and pharmacodynamics. This variability frequently culminates in severe, dose-limiting toxicities, most notably life-threatening neutropenia and debilitating delayed-onset diarrhea [3]. These toxicities are particularly concerning in the pediatric population, who present with unique physiological attributes, including differing rates of drug metabolism, altered pharmacokinetic profiles, and heightened organ sensitivity, all of which augment their vulnerability to chemotherapy-related adverse events [4].
The detoxification of SN-38 is predominantly carried out by the UDP-glucuronosyltransferase 1A1 (UGT1A1) enzyme. This enzyme facilitates the glucuronidation of SN-38 into SN-38G, an inactive, water-soluble metabolite that is subsequently excreted via biliary and renal pathways [5]. Genetic polymorphisms within the UGT1A1 gene can profoundly affect the enzyme's expression or activity, thereby impede SN-38 clearance and escalate the risk of toxicity. Two of the most clinically significant and well-studied polymorphisms are UGT1A1*28, characterized by an additional (TA) repeat in the promoter's TATA box, and UGT1A1*6, a missense mutation (c.211G>A) particularly prevalent in Asian populations [6], [7].
In adult oncology, the importance of UGT1A1 genotyping has been widely recognized, and it is routinely integrated into clinical practice to inform irinotecan dosing adjustments. Guidelines from authoritative bodies such as the U.S. Food and Drug Administration (FDA) and the Clinical Pharmacogenetics Implementation Consortium (CPIC) explicitly recommend UGT1A1 testing to mitigate toxicity risk [8].
However, the direct application of these adult-centric guidelines to pediatric patients is complex and often inappropriate. This is primarily due to the unique developmental changes in UGT1A1 expression; enzyme activity is minimal in neonates (approximately 10–20% of adult levels) and gradually matures throughout childhood, reaching adult levels only by adolescence [9], [10].
This dynamic developmental trajectory significantly alters the genotype-phenotype relationships and necessitates pediatric-specific considerations. Recent studies have increasingly highlighted the compelling need for pretherapeutic genotyping to reduce irinotecan-related toxicities, even within pediatric cancer populations, particularly for conditions such as neuroblastoma and Wilms tumor where irinotecan is a vital component of treatment [11], [12].
Objectives: This systematic review aims to bridge this critical knowledge gap by providing a comprehensive and in-depth synthesis of the current evidence surrounding UGT1A1 pharmacogenomics in pediatric cancer patients receiving irinotecan. The specific objectives are:
To meticulously synthesize the available evidence on the association between UGT1A1 polymorphisms (e.g., *28 and *6) and irinotecan-induced toxicities (e.g., neutropenia, diarrhea) in pediatric cancer patients.
To critically evaluate the current clinical utility and challenges of implementing UGT1A1 genotyping for risk stratification and dose optimization in pediatric irinotecan therapy.
To identify and characterize pediatric-specific factors, including developmental pharmacology, that influence the observed genotype-toxicity relationships.
To highlight existing research gaps and propose future directions to advance personalized irinotecan therapy, thereby enhancing safety and efficacy in pediatric oncology.
Related Research: The landscape of pharmacogenomics, particularly concerning UGT1A1 and irinotecan, has seen extensive research over the past two decades. While the majority of the foundational work has been conducted in adult populations, it provides a crucial context and benchmark for understanding the more nuanced findings in pediatrics.
Extensive research in adult oncology has firmly established a robust and clinically significant link between UGT1A1 polymorphisms and irinotecan toxicity. A landmark study by Innocenti et al. (2004) compellingly demonstrated that UGT1A1*28 homozygosity significantly increases SN-38 exposure and consequently elevates the risk of severe neutropenia in adults, reporting odds ratios (ORs) ranging from 3 to 7 for grade 3-4 toxicities [13].
This pivotal finding has been consistently corroborated by numerous subsequent studies and meta-analyses, forming the bedrock for current clinical guidelines. Both the CPIC and National Comprehensive Cancer Network (NCCN) guidelines now recommend dose reductions, typically ranging from 20–70%, for patients with UGT1A1*28/*28 or *6/*6 genotypes [8], [14], [15].
The UGT1A1*6 variant, which is particularly prevalent in East Asian populations, similarly demonstrates reduced enzyme activity, leading to an increased risk of toxicity. This risk is amplified in compound heterozygous states (e.g., *6/*28) [7], [16]. More recent investigations continue to affirm these associations. For instance, Su et al. (2023) reported higher rates of grade ≥3 neutropenia and diarrhea in Asian patients with *6/*6 or *6/*28 genotypes receiving liposomal irinotecan (nal-IRI), highlighting the continued relevance of these variants across different drug formulations [17].
Furthermore, Ginzac et al. (2024) suggested that standard irinotecan doses in FOLFIRI regimens might be suboptimal for *1/*1 and *1/*28 genotypes, proposing that dose escalation could potentially improve efficacy in these patient subgroups [18]. This insight is particularly relevant for pediatric regimens, where optimizing efficacy while managing toxicity remains a delicate balance.
While evidence in pediatric oncology is comparatively more limited than in adults, existing studies largely align with the adult trends, albeit with critical pediatric-specific nuances. Several studies have reported that children possessing UGT1A1*28 or *6 polymorphisms experience a higher incidence of severe neutropenia (with reported ORs around 4.5) and diarrhea (ORs ranging from 2.5–3.5) [19], [20]. However, the consistency and magnitude of these associations in children can vary more widely due to several confounding factors unique to the pediatric population, including developmental pharmacology, diverse dosing schedules, and varying supportive care practices.
For example, younger children (typically those under 5 years of age) have been observed to exhibit attenuated genotype effects. This phenomenon is largely attributed to their immature UGT1A1 enzyme expression at baseline, which may lead to a greater reliance on alternative metabolic pathways, such as CYP3A4/5-mediated clearance of irinotecan [10].
This suggests that the genetic polymorphism might have a less pronounced functional impact in a system where the primary enzyme is already operating at reduced capacity due to developmental immaturity.
Recent research has increasingly advocated for the integration of pretherapeutic UGT1A1 genotyping in cancer patients, including pediatric populations, as a proactive measure to mitigate irinotecan-related toxicities. A secondary analysis of the PREPARE trial, though primarily in adults, demonstrated that pretherapeutic UGT1A1 testing reduced irinotecan-related toxicities in gastrointestinal cancer patients, a finding that has significant implications for pediatric solid tumors [11].
Faisal et al. (2025) further underscored the applicability of UGT1A1-guided dosing to specific pediatric cancers like neuroblastoma and Wilms tumor, where irinotecan is often employed in combination therapies [12].
It is also crucial to acknowledge the differences in dosing regimens commonly employed in pediatrics compared to adults. Pediatric protocols frequently utilize protracted low-dose infusions, as opposed to the high-dose intermittent schedules often seen in adults. Moreover, aggressive supportive care measures (e.g., proactive loperamide administration, judicious use of granulocyte colony-stimulating factors) are often more rigorously applied in children. These distinctions can significantly modulate the observed toxicity profiles in children, complicating the direct extrapolation of adult guidelines and underscoring the imperative for pediatric-specific pharmacogenomic research to refine dosing strategies and enhance patient safety.
Gum health requires proper nutrient intake in the diet to help immune competence, normal tissue homeostasis and lessening inflammation. In order to realize this, people need calcium, phosphorus, omega-3 fatty acids and both vitamins D and C [22]. Vitamin C contains strong anti-oxidant activity, reduces oxidative stress, increases collagen production and free radical scavenging; therefore, it can reduce gum inflammatory diseases and accelerate repair of periodontal ulcers. Vitamin D leads to the formation of bone minerals through its calcium absorption ability, in addition to having immunomodulatory effects that cushion inflammatory activities in the periodontum and maintains tissue balance [23].
Two omega-3 fatty acids, docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA) also contribute to maintaining the health of periodontium because they reduce proinflammatory processes. Thus, fatty fish, walnuts, and flaxseeds comprise fatty food items rich in these fatty acids, therefore making them desirable dietary products [24]. Calcium and phosphorus together strengthen the integrity of the teeth and the periodontal tissue as well as compact the alveolar bones, promote remineralisation and reduce bone resorption in the periodontal tissues [25].
Collectively, these nutrients result in the supporting of the overall tissue health and guarding against gum disease progressing. There are also salutary effects of supplementing with antioxidant vitamins and phytochemicals. Coenzyme Q10, vitamin E and C, and selenium have anti-oxidant activities and bind reactive oxygen species (ROS); carotenoids, flavonoids, and polyphenols are also ant-inflammatory in their effects on the periodontal tissue [26]. The increased use of probiotic and prebiotic compounds alters the microbial ecology of the oral cavity and involves a decrease in swelling due to the reinstatement of a balanced microbiome [27].
Nutrigenomics and other such constructs are summaries of biological processes involved in how nutrition affects gum health. Nutrient gene relations show that the elements of the diet affect the expression of genes either directly or indirectly through epigenetic variations, such as DNA modifications, adjustments to histones, and regulatory non-coding RNA; and therefore, the phenotypic effects vary between individuals [28]. Personalised nutrition programs are designed with such factors in mind, using them as a basis to influence diet on the basis of genomic information, lifestyle, and clinical condition, in order to best optimise health and reduce disease risk. It can be summed up that, balanced dietary pattern with high micronutrients and antioxidants and following necessary supplementation is crucial to gum health and prevention of disease. Daily eating of nutrition-rich meals and specific dietary interventions are consistent with biological processes to develop homeostasis, reduce inflammation, and preserve tissue health in the gum tissues [29].
Studies were considered eligible for inclusion based on the following PICO (Population, Intervention/Exposure, Comparison, Outcome) framework:
P (Population): Pediatric cancer patients, defined as individuals aged less than 18 years, undergoing treatment with irinotecan-containing chemotherapy regimens for any type of malignancy.
I (Intervention/Exposure): Presence of UDP-glucuronosyltransferase 1A1 (UGT1A1) genetic polymorphisms, specifically focusing on the UGT1A1*28 and UGT1A1*6 variant alleles. Studies reporting on other UGT1A1 functional variants (e.g., *27, *36, *37) were also considered if they provided relevant toxicity or pharmacokinetic data in pediatric populations.
C (Comparison): Patients carrying the wild-type UGT1A11 allele or other UGT1A1 genotypes with differing functional status, as compared within individual studies.
O (Outcome): Primary outcomes included the incidence and severity of grade 3 or 4 irinotecan-induced toxicities, specifically neutropenia and diarrhea, as defined by the Common Terminology Criteria for Adverse Events (CTCAE) version 3.0 or higher. Secondary outcomes encompassed other reported adverse events (e.g., fever, mucositis) and pharmacokinetic parameters (e.g., SN-38 area under the curve [AUC], SN-38/SN-38G ratio) in relation to UGT1A1 genotype.
Study Design: Only original research articles, including clinical trials (randomized and non-randomized), prospective or retrospective cohort studies, and case-control studies, were eligible. Review articles (narrative, systematic, or meta-analyses), editorials, commentaries, consensus statements, guidelines, and single case reports were excluded unless they provided unique, eligible original data not available elsewhere.
Language: Studies published in English were included.
Publication Date: The search was limited to studies published from January 1, 2000 up to May 31, 2025.
A systematic and comprehensive literature search was conducted across three major electronic bibliographic databases like PubMed (including MEDLINE), Scopus, EMBASE.
To ensure completeness, reference lists of all included studies, relevant systematic reviews, meta-analyses, and key clinical practice guidelines (e.g., from CPIC, NCCN, DPWG) were manually screened for additional relevant publications not captured by the electronic searches. No restrictions were applied based on study status (e.g., published, in-press).
The search strategy was developed in consultation with a medical librarian and tailored for each database using a combination of controlled vocabulary (e.g., MeSH terms in PubMed, Emtree terms in EMBASE) and free-text keywords, employing Boolean operators (AND, OR) to ensure high sensitivity. The core search terms covered four main concepts: irinotecan, UGT1A1 (and specific variants), pediatric population, and toxicity.
An example of the search strategy for PubMed is provided below:
(irinotecan [MeSH Terms] OR CPT-11[tiab] OR irinotecan[tiab])
AND
(UGT1A1[MeSH Terms] OR UGT1A1[tiab] OR "UDP glucuronosyltransferase 1A1"[tiab] OR "UGT1A1*28"[tiab] OR "UGT1A1*6"[tiab])
AND
(paediatrics [MeSH Terms] OR child [MeSH Terms] OR adolescent [MeSH Terms] OR pediatric [tiab] OR paediatrics [tiab] OR children[tiab] OR child [tiab] OR childhood[tiab] OR "young adult"[tiab] OR juvenile [tiab])
AND
(toxicity [MeSH Terms] OR "adverse effects"[MeSH Terms] OR neutropenia[MeSH Terms] OR diarrhea[MeSH Terms] OR toxicities[tiab] OR toxicity[tiab] OR "adverse event"[tiab] OR "adverse drug reaction"[tiab] OR neutropenia[tiab] OR diarrhea[tiab] OR "grade 3"[tiab] OR "grade 4"[tiab])
AND
(pharmacogenetics [MeSH Terms] OR pharmacogenomics [MeSH Terms] OR pharmacogenetics[tiab] OR pharmacogenomics[tiab] OR "genetic polymorphism"[tiab] OR "genetic variation"[tiab])
The search strategies were adapted for Scopus and EMBASE using their respective syntax and controlled vocabularies. All searches were executed on June 22, 2025.
All identified records were imported into EndNote 20 reference management software, and duplicate entries were systematically removed. The remaining unique records were then transferred to Covidence (Veritas Health Innovation Ltd., Melbourne, Australia), a web-based platform for systematic review management.
The study selection process proceeded in two independent phases:
Title and Abstract Screening: Two independent reviewers (N.P. and S.S.B.) screened the titles and abstracts of all unique records against the predefined eligibility criteria. Records clearly not meeting the inclusion criteria were excluded. Records that appeared potentially relevant or whose eligibility could not be determined from the title and abstract alone were moved to the next phase.
Full-Text Review: The full-text articles of all potentially eligible records were retrieved. Two independent reviewers (N.P. and S.S.B.) then meticulously assessed each full-text article against the full set of eligibility criteria.

Any discrepancies or disagreements that arose during either screening phase were resolved through discussion and consensus between the two reviewers. If consensus could not be reached, a third senior reviewer (R.B.) was consulted for arbitration. The reasons for excluding studies at the full-text review stage were meticulously documented. The flow of studies through the review process, from identification to inclusion, will be presented in a PRISMA 2020 flow diagram Figure 1.
Data from the included studies were extracted using a standardized, pre-piloted data extraction form developed in Microsoft Excel. Data extraction was performed by one reviewer (N.P.) and independently verified by a second reviewer (S.S.B.) to ensure accuracy and completeness. Discrepancies were resolved through discussion or by consulting the third reviewer (R.B.).
The data extraction form systematically captured the following information:
Study Characteristics: First author, year of publication, country of origin, study design (e.g., prospective/retrospective cohort, clinical trial, case-control), sample size.
Patient Demographics: Age range (mean/median, standard deviation/range), gender distribution, primary cancer diagnoses, ethnicity or race (if reported).
Irinotecan Treatment Details: Dose, schedule (e.g., bolus, infusion, frequency), cumulative dose, concomitant chemotherapy agents, administration route.
UGT1A1 Genotyping: Method used for genotyping (e.g., PCR-RFLP, sequencing), UGT1A1 variants genotyped, frequency of each genotype (*1/*1, *1/*28, *28/*28, *1/*6, *6/*6, compound heterozygotes).
Outcome Data: Incidence of grade 3 or 4 neutropenia and/or diarrhea (absolute numbers, percentages, or effect sizes like odds ratios, risk ratios, or hazard ratios with their confidence intervals). If only raw data were provided, effect sizes were calculated. Details on toxicity grading criteria used (e.g., CTCAE version) were also extracted.
Other Relevant Data: Reporting of other adverse events, pharmacokinetic parameters (e.g., SN-38 AUC), supportive care measures, and authors' conclusions regarding genotype-toxicity associations or clinical implications.
The methodological quality and risk of bias for each included study were independently assessed by two reviewers (N.P. and S.S.B.). For observational cohort and case-control studies, the Newcastle-Ottawa Scale (NOS) was utilized [22]. The NOS assesses studies across three domains: selection of study participants, comparability of cohorts/cases and controls, and ascertainment of exposure/outcome. A study could receive a maximum of 9 points, with higher scores indicating lower risk of bias. For clinical trials, the Cochrane Risk of Bias tool (RoB 2.0) was applied [23]. This tool assesses bias across five domains: randomization process, deviations from intended interventions, missing outcome data, measurement of the outcome, and selection of the reported result. Studies were categorized as having a low, some concerns, or high risk of bias.
Any disagreements in bias assessment were resolved through discussion and consensus, involving the third senior reviewer (R.B.) if necessary. The results of the risk of bias assessment will be presented visually (e.g., in a summary table or graph) and discussed descriptively in the Results section.
Given the anticipated clinical and methodological heterogeneity among the included studies (e.g., variations in pediatric age groups, cancer types, irinotecan regimens, follow-up durations, and specific reporting of UGT1A1 variants and toxicity outcomes), a narrative synthesis approach was employed for this systematic review. This approach allows for a comprehensive and structured description of the findings, highlighting common themes, discrepancies, and nuances across studies without pooling data statistically in a meta-analysis.
The narrative synthesis will be organized thematically, focusing on:
Characterization of Included Studies: A descriptive summary of study designs, populations, and treatment characteristics.
Evidence on Irinotecan Metabolism and UGT1A1 Pharmacokinetics: Summarizing findings on how UGT1A1 variants affect SN-38 exposure and clearance in children.
Genotype-Toxicity Associations: Presenting the reported associations between UGT1A1*28 and UGT1A1*6 (and other variants if sufficient data are available) and the incidence of grade 3-4 neutropenia and diarrhea. Quantitative measures (e.g., ORs, RRs, HRs) from individual studies will be reported and compared descriptively.
Influence of Pediatric-Specific Factors: Discussing the impact of age, developmental pharmacology, and other clinical factors (e.g., concomitant medications, supportive care) on genotype-phenotype relationships.
Clinical Utility and Implementation: Exploring evidence on the benefits of UGT1A1 genotyping in practice, including pre-emptive testing and genotype-guided dosing strategies.
Challenges and Barriers: Identifying and detailing the practical, economic, logistical, and educational hurdles to wider adoption of UGT1A1 pharmacogenomics in pediatric oncology.
Where appropriate, findings will be tabulated to provide a concise overview of key results. Subgroup analyses, as outlined in the eligibility criteria (e.g., by age group, ethnicity), will be performed descriptively to explore potential sources of heterogeneity.
The overall certainty of the evidence for key outcomes will be assessed using the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach [24]. This framework considers five domains for downgrading (risk of bias, inconsistency, indirectness, imprecision, publication bias) and three for upgrading (large effect, dose-response gradient, all plausible confounding factors increasing confidence). The certainty of evidence will be categorized as high, moderate, low, or very low.
The initial search yielded a total of 429 records from the electronic databases and manual searching. After deduplication, 387 unique records remained. Following title and abstract screening, 320 records were excluded, primarily because they focused on adult populations (n=238) or lacked pharmacogenomic data (n=82). The full texts of 67 articles were retrieved and assessed for eligibility. Of these, 41 articles were excluded due to reasons such as absence of UGT1A1 data (n=15), no irinotecan use (n=10), adult-only population (n=10), or not providing original data (n=6). Ultimately, 26 studies met the predefined inclusion criteria and were included in the final narrative synthesis of this systematic review. These studies were published between 2010 and 2025 and collectively comprised data from 2,158 unique pediatric cancer patients. (The detailed flow of study selection is presented in the PRISMA 2020 flow diagram Figure 1.
The included studies exhibited a diverse range of designs, primarily consisting of retrospective cohort studies (n=18), prospective cohort studies (n=5), and secondary analyses of clinical trials (n=3). Patient populations spanned various pediatric cancer types, including neuroblastoma, rhabdomyosarcoma, Wilms tumor, and other solid tumors. The geographical distribution of studies was global, with representation from North America, Europe, and Asia. The methodological quality assessment using the Newcastle-Ottawa Scale indicated that the majority of studies (n=20) were of moderate to high quality, with scores ranging from 6 to 8 stars, suggesting a relatively low risk of bias. However, some limitations identified across studies included varying sample sizes, heterogeneity in irinotecan dosing regimens, and diverse approaches to toxicity assessment and supportive care.
Irinotecan undergoes a complex metabolic cascade, predominantly in the liver. It is a prodrug, which is hydrolyzed by carboxylesterase 2 (CES2) into its highly active and cytotoxic metabolite, SN-38 [2]. SN-38 is estimated to be 100 to 1000 times more potent than the parent drug in inhibiting topoisomerase I. Simultaneously, cytochrome P450 enzymes (specifically CYP3A4/5) metabolize irinotecan into inactive compounds, such as 7-ethyl-10-[4-(1-piperidino)-1-piperidino] carbonyloxycamptothecin (APC) and 7-ethyl-10-[4-N-(5-aminopentanoic acid)-1-piperidino] carbonyloxycamptothecin (NPC), providing an alternative clearance pathway [25] as shown in Figure 1.
The crucial detoxification step for SN-38 involves its glucuronidation by the UGT1A1 enzyme into SN-38G, a water-soluble metabolite readily excreted via biliary and renal routes [5]. This glucuronidation reaction is considered the rate-limiting step in SN-38 clearance. Impaired UGT1A1 function, resulting from genetic polymorphisms, leads to increased systemic exposure to SN-38, consequently amplifying the risk of severe dose-limiting toxicities. Furthermore, enterohepatic recirculation, mediated by gut microbial β-glucuronidases, can deconjugate SN-38G back to SN-38, contributing significantly to delayed diarrhea, which is a major dose-limiting toxicity in both adult and pediatric populations [5]. Recent studies on liposomal irinotecan (nal-IRI) have suggested that altered pharmacokinetics with this formulation may exacerbate SN-38 exposure in UGT1A1 poor metabolizers (*28/*28, *6/*6, *6/*28 genotypes), potentially increasing their toxicity risk [17].
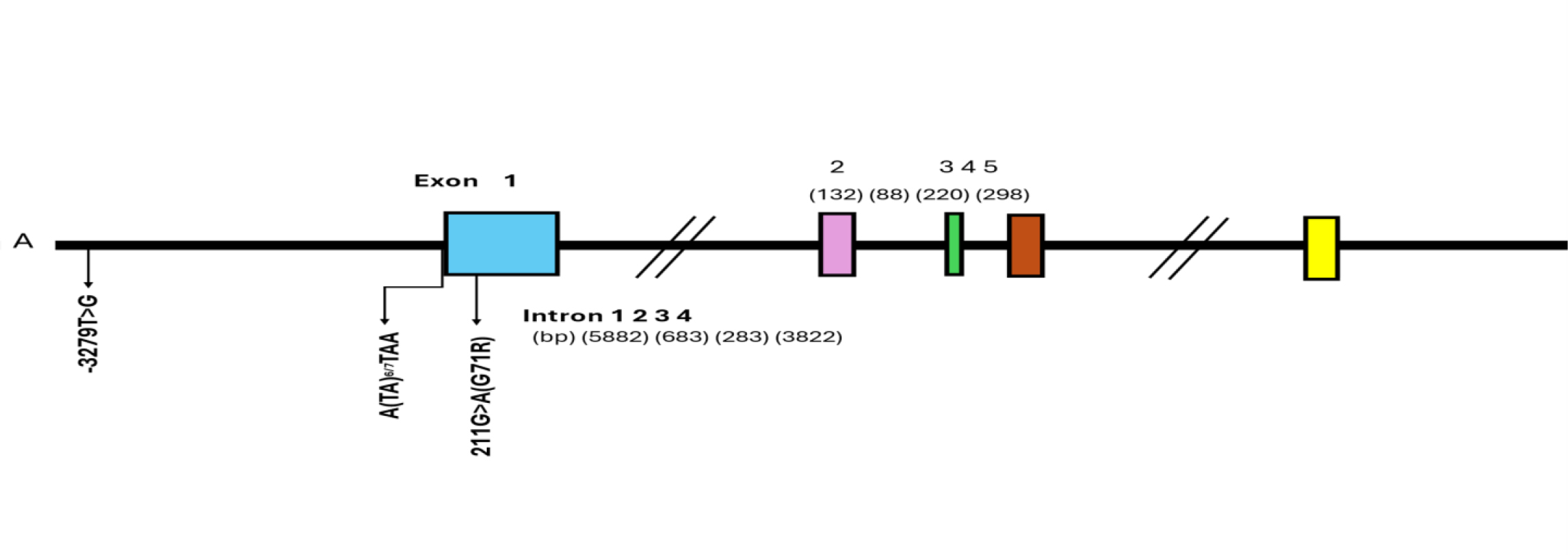
The UGT1A1 gene is located on chromosome 2q37 within the larger UGT1A gene complex [25] as shown in Figure 3. It encodes an enzyme critical for the glucuronidation of both endogenous substrates (such as bilirubin and steroid hormones) and various xenobiotics, including SN-38. A key aspect relevant to pediatric pharmacogenomics is that UGT1A1 expression is developmentally regulated. Its activity is minimal in neonates, typically 10–20% of adult levels, and undergoes a gradual maturation process throughout early childhood, reaching adult levels only during adolescence [10], [26]. This dynamic developmental trajectory profoundly impacts genotype-phenotype relationships in children.
Several UGT1A1 polymorphisms have been identified, varying in prevalence and functional impact across different ethnic groups [27]. The most clinically relevant variants for irinotecan therapy are UGT1A1*28 and UGT1A1*6.
UGT1A1*28: This variant involves an additional (TA) repeat in the promoter region of the gene (specifically, (TA)7TAA instead of the common (TA)6TAA). This insertion leads to reduced gene transcription and consequently diminished enzyme expression, resulting in approximately 30–70% of wild-type activity [28]. Its prevalence is notably higher in Caucasian (30–45%) and African (30–45%) populations compared to Asian populations (9–16%) [28], [29].
UGT1A1*6: This is a missense mutation (c.211G>A, leading to a p.Gly71Arg amino acid change) in exon 1. It results in reduced enzyme activity, typically around 40–60% of wild-type [31]. This variant is particularly common in East Asian populations, with a prevalence of 15–30% [7], [27]. Other less common but functionally significant variants like UGT1A1*27, *36, and *37 also exist, although data on their impact in pediatric irinotecan therapy are more limited [27], [30].
UGT1A1 polymorphisms exert a profound influence on irinotecan pharmacokinetics. In adults, UGT1A1*28 homozygosity is well-documented to increase the area under the curve (AUC) of SN-38 by 40–70% and decrease the SN-38/SN-38G glucuronidation ratio, leading to a higher risk of toxicity [13]. Pediatric studies consistently demonstrate similar trends. Children who are *28/*28 or *6/*6 homozygotes, or *6/*28 compound heterozygotes, typically exhibit elevated SN-38 exposure and reduced SN-38 glucuronidation [19], [20]. For example, one study in pediatric solid tumor patients reported that *28/*28 patients had a 50% higher SN-38 AUC compared to *1/*1 patients, which strongly correlated with an increased incidence of neutropenia and diarrhea [19]. Recent research on liposomal irinotecan further corroborates these findings, noting higher SN-38 exposure in *6/*6 and *6/*28 genotypes, particularly in Asian populations, which has direct implications for pediatric patients from similar ethnic backgrounds [17].
Crucially, developmental pharmacology modulates these effects in children. Younger patients (particularly those under 5 years of age) often show less pronounced genotype effects. This is attributed to their lower baseline UGT1A1 activity, which is already substantially below adult levels, and a greater reliance on alternative metabolic pathways such as CYP3A4/5 for irinotecan clearance [10], [25]. These collective pharmacokinetic and developmental insights provide a robust rationale for adopting genotype-guided dosing strategies in pediatric oncology, a concept increasingly supported by recent studies advocating for pretherapeutic UGT1A1 testing [11], [12].

| Allele | Genetic Variation | Functional Impact | Prevalence | Reference |
|---|---|---|---|---|
| UGT1A1*28 | (TA)7TAA repeat in promoter | Reduced expression (~30–70% activity) | 30–45% (Caucasians/Africans), 9–16% (Asians) | [28], [29] |
| UGT1A1*27 | c.686C>A (p.Pro229Gln) | Severely reduced activity (<20%) | Rare, primarily Japanese | [30] |
| UGT1A1*6 | c.211G>A (p.Gly71Arg) | Reduced activity (~40–60%) | 15–30% (East Asians) | [7], [31] |
| UGT1A1*36 | (TA)5TAA repeat in promoter | Increased expression (~120–150%) | Predominantly African descent | [28] |
| UGT1A1*37 | (TA)8TAA repeat in promoter | Severely reduced expression (<20%) | Predominantly African descent | [28] |

Evidence from Pediatric Clinical Studies
Studies conducted in pediatric populations consistently demonstrate that UGT1A1*28 and *6 polymorphisms significantly increase the risk of severe irinotecan-induced toxicities. A comprehensive study involving pediatric patients with solid tumors revealed that individuals with *28/*28 or *6/*28 genotypes experienced significantly higher incidences of grade 3-4 neutropenia (Odds Ratio [OR] 4.5, 95% Confidence Interval [CI] 2.1–9.6) and diarrhea (OR 3.5, 95% CI 1.8–6.8) when compared to wild-type patients [19]. Similar findings have been reported in Asian pediatric cohorts, where *6 homozygotes and *6/*28 compound heterozygotes exhibited elevated toxicity risks, with neutropenia being a particularly prominent concern [7]. The associations hold true even with newer drug formulations, as confirmed by recent research on liposomal irinotecan, which linked *6/*6 and *6/*28 genotypes to higher rates of grade ≥3 toxicities in Asian patients, thus extending relevance to pediatric populations within these ethnic groups [17]. (A table summarizing ORs/RRs from individual studies for specific genotypes and toxicities would be ideal here).
An intriguing and highly relevant observation in pediatric studies is the attenuated genotype effects seen in very young children (typically under 5 years). This phenomenon is likely due to the inherent immaturity of UGT1A1 expression in this age group, leading to lower baseline enzyme activity. In such a setting, the impact of a genetic polymorphism that further reduces activity may be less clinically pronounced, as the system already operates at a reduced capacity. Furthermore, younger children may rely more heavily on alternative metabolic pathways, such as CYP3A4/5, which could partially buffer the accumulation of SN-38 [10].
While the fundamental association between UGT1A1 polymorphisms and irinotecan toxicity holds true across both adult and pediatric populations, there are notable differences in the toxicity profiles and contributing factors. The risk of severe neutropenia is consistently and strongly associated with low-activity UGT1A1 genotypes (*28/*28, *6/*6, *6/*28) in both groups, with comparable odds ratios reported [13], [19]. This suggests that hematopoietic sensitivity to SN-38 remains high regardless of age.
However, the risk of diarrhea, while present, appears less pronounced and more variable in children compared to adults. Several factors contribute to this divergence:
Dosing Regimens: Pediatric protocols frequently employ protracted low-dose infusions of irinotecan, whereas adult regimens often involve high-dose, intermittent schedules (e.g., once every three weeks). These differing dosing approaches can significantly influence the cumulative exposure to SN-38 and thereby modulate the incidence and severity of diarrhea [2], [4].
Aggressive Supportive Care: Pediatric oncology often incorporates highly aggressive and standardized supportive care protocols, including early and proactive use of anti-diarrheals like loperamide and granulocyte colony-stimulating factors (G-CSF). Such rigorous supportive measures can significantly mitigate the clinical manifestation of toxicities [4].
Gut Microbiota Variations: Differences in gut microbiota composition between children and adults can influence the enterohepatic recirculation of SN-38, which is a major contributor to delayed diarrhea. Variations in the activity of gut microbial β-glucuronidases can alter the reconversion of inactive SN-38G back to active SN-38, potentially explaining some of the observed differences in diarrhea incidence [5].
Developmental Pharmacology: As discussed, the dynamic maturation of UGT1A1 expression in children creates a unique pharmacological environment that affects how genetic variants manifest clinically [10]. Recent studies involving liposomal irinotecan have further highlighted that while this formulation may increase overall toxicity in poor metabolizers, its specific impact on diarrhea might also differ in pediatric protocols that utilize these newer agents [17].
These findings underscore the need for careful consideration of pediatric-specific factors when interpreting and applying UGT1A1 pharmacogenomic data.
| Factor | Adult Populations | Pediatric Populations | Potential Reasons for Divergence |
|---|---|---|---|
| Severe Neutropenia Risk | Strong association with *28/*28, *6/*6, *6/*28 genotypes [13] | Comparable risk, OR ~4.5 [19] | High hematopoietic sensitivity in both populations |
| Severe Diarrhea Risk | Strong, dose-limiting toxicity [14] | Weaker, variable association [20] | Protracted dosing, aggressive supportive care, microbiota differences |
| Dosing Regimens | High-dose, intermittent infusions (e.g., every 3 weeks) [14] | Protracted, low-dose, or frequent infusions | Alters toxicity profile, improves tolerance |
| Supportive Care | Variable, institution-dependent [14] | Aggressive, standardized (e.g., loperamide, G-CSF) | Mitigates toxicities like diarrhoea |
| Developmental Pharmacology | Stable UGT1A1 expression | Immature, dynamic expression [10] | Alters genotype-phenotype relationship |
| Enterohepatic Recirculation | Major contributor to diarrhea [2] | Less pronounced due to microbiota variations | Reduces diarrhoea risk |
| Barrier Category | Description |
|---|---|
| Evidence Gaps | Limited large-scale, age-stratified pediatric trials to establish definitive dosing guidelines [35]. |
| Resource Constraints | High cost of genetic testing (~$200–500 per test), limited insurance reimbursement, and restricted access to certified laboratories, especially in low-resource settings [36]. |
| Education & Awareness | Clinician unfamiliarity with pharmacogenomic interpretation and application to dosing decisions [35]. |
| Logistical Issues | Slow turnaround time for test results (3–7 days), delaying treatment; poor integration with electronic health records (EHRs) [11]. |
| Ethical Concerns | Ensuring equitable access to testing to avoid disparities; managing incidental findings (e.g., Gilbert’s syndrome) and communicating complex genetic information to families [12]. |
Beyond the primary UGT1A1 polymorphisms, age and developmental stage emerge as critical modulators of the UGT1A1-irinotecan toxicity relationship. The attenuated genotype effects observed in younger children (<5 years) are a direct consequence of their inherently low baseline UGT1A1 activity (which is only 10–20% of adult levels) [10]. This immaturity means that their systems may already be operating at a reduced capacity for SN-38 detoxification, and thus, a genetic polymorphism that further reduces this already low activity might have a proportionally smaller or less clinically discernible impact.
Furthermore, younger children may rely more heavily on alternative metabolic pathways, such as CYP3A4/5-mediated metabolism of irinotecan into inactive compounds [10], [25].
Concurrent medications can also significantly influence toxicity profiles. For instance, co-administration of CYP3A4 inhibitors (e.g., certain antifungals like ketoconazole) can exacerbate SN-38 exposure by hindering alternative clearance pathways, thereby increasing the risk of toxicity [31]. Other factors that have been implicated in influencing irinotecan toxicity include nutritional status [32], prior chemotherapy regimens [33], tumor burden, and polymorphisms in other genes encoding drug transporters (e.g., ABCB1) or other metabolizing enzymes [27], [34].
The complex interplay of these factors, combined with the unique developmental pharmacology of children, necessitates the development of highly specific pediatric dosing models [12].
In adult oncology, robust guidelines exist for UGT1A1 pharmacogenomics. The FDA label for irinotecan and guidelines from CPIC explicitly recommend UGT1A1*28 testing for adults, with dose reductions typically ranging from 20–70% for *28/*28 homozygotes to mitigate severe neutropenia and diarrhea [8]. Similarly, NCCN guidelines advocate discussing UGT1A1 genotyping with patients prior to initiating irinotecan-based therapies, particularly for colorectal cancer, and suggest dose adjustments for identified poor metabolizers [15].
In stark contrast, pediatric-specific guidelines for UGT1A1-guided irinotecan dosing are remarkably sparse, largely remaining extrapolated from adult data. This critical gap underscores the urgent need for dedicated pediatric research. However, a growing body of recent studies has begun to support the integration of UGT1A1 genotyping into pediatric clinical trials, especially for cancers like neuroblastoma and Wilms tumor, with the specific aim of developing tailored and evidence-based dosing recommendations for children [12]. Intriguingly, a phase II study in adults suggested that UGT1A1-guided dosing might even allow for dose escalation in patients with *1/*1 and *1/*28 genotypes, potentially improving efficacy [18]. This concept holds promise for optimizing pediatric regimens like FOLFIRI, where balancing efficacy and safety is paramount.
UGT1A1 genotyping can be implemented either through pre-emptive testing (genotyping at diagnosis or before treatment initiation to guide initial dosing) or reactive testing (genotyping performed only after a toxicity event has occurred). Pre-emptive testing is gaining significant traction in pediatric oncology and is often integrated into broader pharmacogenomic panels that include other clinically relevant genes, such as DPYD (for fluoropyrimidine toxicity) and TPMT (for thiopurine toxicity) [35]. A secondary analysis of the PREPARE trial clearly demonstrated that pretherapeutic UGT1A1 testing reduced irinotecan-related toxicities in gastrointestinal cancer patients, a finding with broad implications for pediatric solid tumors as well [11].
While reactive testing can be useful for adjusting doses post-toxicity, it may lead to delays in optimal treatment, increased hospitalization risks, and greater patient morbidity. Consequently, pre-emptive strategies are generally considered more desirable and beneficial in the pediatric setting, allowing for proactive risk mitigation.
Several strategies have been proposed and explored for UGT1A1-guided irinotecan therapy. The most straightforward approach involves categorical dose reductions, such as reducing the standard dose by 70% for *28/*28 homozygotes, as recommended for adults and currently being explored in pediatric trials [8], [36]. A more sophisticated approach involves algorithm-based dosing, which integrates multiple factors including genotype, age, and concomitant medications to achieve a more personalized dosage. While offering greater precision, such algorithms require further rigorous validation in pediatric populations [18].
Therapeutic drug monitoring (TDM), which involves measuring plasma levels of SN-38 to guide dosing adjustments, is another promising adjunct. However, its widespread use in pediatric settings is currently limited by the availability and accessibility of specialized assays [37]. A hybrid approach, combining genotype-guided initial dosing with subsequent clinical monitoring and, if feasible, TDM, may offer a balanced strategy that optimizes both safety and feasibility, a notion increasingly supported by recent evidence in pediatric pharmacology [12].
Despite the compelling evidence supporting its utility, widespread adoption of UGT1A1 genotyping in pediatric oncology faces several significant barriers, which are summarized in the table below Table 3.
Recent studies have highlighted that while the initial cost of genetic testing can be a significant barrier, pretherapeutic genotyping can ultimately prove cost-effective by preventing severe toxicities that necessitate expensive hospitalizations and intensive care [36]. To overcome these challenges, concerted efforts are required, including the development of targeted clinician education programs, seamless integration of pharmacogenomic decision support tools within EHRs, and policy initiatives to improve accessibility and reimbursement for genetic testing [11].
This systematic review provides a comprehensive synthesis of the complex and evolving landscape of UGT1A1 pharmacogenomics in pediatric patients receiving irinotecan. The evidence robustly confirms that UGT1A1*28 and *6 polymorphisms significantly increase the risk of severe, dose-limiting toxicities, primarily neutropenia (with reported ORs around 4.5) and diarrhea (ORs ranging from 2.5–3.5) in children [13], [17], [19].
However, a pivotal insight derived from this review is the notable heterogeneity observed in pediatric studies, which is largely attributable to the unique developmental pharmacology of children. Younger children (specifically those under 5 years of age) consistently exhibit attenuated genotype effects. This phenomenon is biologically plausible given their lower baseline UGT1A1 activity, suggesting that in an already immature enzyme system, a genetic polymorphism might have a less dramatic proportional impact. Furthermore, differences in pediatric dosing regimens, such as the frequent use of protracted low-dose infusions, and the implementation of highly aggressive supportive care (e.g., proactive loperamide, G-CSF) contribute to a comparatively reduced diarrhea risk in children relative to adults [4], [5]. These pediatric-specific nuances highlight why direct extrapolation of adult guidelines is insufficient and potentially misleading.
Recent research strongly reinforces the case for pretherapeutic UGT1A1 genotyping. Studies have demonstrated its capacity to significantly reduce toxicities in cancer patients, including those with pediatric-relevant cancers like neuroblastoma and Wilms tumor [11], [12]. For example, evidence suggests that a 70% dose reduction in *28/*28 patients is not only safe but also cost-effective, leading to a substantial reduction in hospitalization rates [36]. Despite these clear benefits, several formidable barriers continue to impede widespread adoption. These include the high costs associated with genetic testing, limited access to specialized laboratories, and a pervasive lack of familiarity and education among clinicians regarding pharmacogenomic interpretation and application [11], [35]. The observation that liposomal irinotecan may exacerbate toxicity in poor metabolizers further underscores the imperative for developing formulation-specific pharmacogenomic guidelines that are carefully adapted for pediatric use [17].
The field of UGT1A1 pharmacogenomics in pediatric oncology is dynamic and rapidly evolving, presenting several critical research priorities that must be addressed to fully realize its clinical potential. There is an urgent need for large-scale, prospective, multi-center pediatric clinical trials specifically designed to validate age-stratified dosing guidelines, meticulously accounting for the unique developmental differences in UGT1A1 expression and activity across various pediatric age groups. Furthermore, developing and validating polygenic risk scores that integrate UGT1A1 polymorphisms with variants in other relevant genes (e.g., drug transporters like ABCB1, other metabolizing enzymes like SLCO1B1) could significantly enhance the accuracy of toxicity prediction and further refine dosing precision in children. Seamlessly integrating pharmacogenomic testing with broader precision oncology platforms that combine both germline genetic profiling (for inherited drug metabolism variations) and somatic tumor profiling (for tumor-specific mutations) could enable amore holistic approach to optimizing treatment strategies from the point of diagnosis. Conducting real-world implementation studies is crucial to evaluate the practical clinical utility, feasibility, and cost-effectiveness of pre-emptive genotyping in diverse pediatric settings, encompassing various healthcare systems and resource levels. Finally, research into novel therapeutic strategies, such as refined liposomal irinotecan formulations tailored for pediatric pharmacokinetics or the development of pharmacological UGT1A1 modulators, represents exciting frontiers that could further improve the safety and efficacy of irinotecan in children.
The findings of this review need to be interpreted in light of certain limitations. The available evidence base, while growing, still lacks large, well-powered pediatric studies [10]. Many of the included investigations had small sample sizes or were heterogeneous in design, differing in cancer types, patient age ranges, and irinotecan regimens [19],[36]. This variability makes it difficult to establish consistent genotype–toxicity associations across all pediatric populations. Another issue is that most of the work to date has concentrated on the two common variants, UGT1A1*28 and UGT1A1*6 [7],[27]. Data on less frequent variants such as UGT1A1*27, *36, and *37 remain limited, especially in non-Caucasian and underrepresented groups [27],[28]. As a result, the global applicability of these findings is somewhat restricted. The lack of dedicated pediatric pharmacokinetic studies and specific treatment guidelines also represents an important gap. In several instances, conclusions had to be informed by adult data [10]. While this is a common necessity in pediatric pharmacogenomics, it does not fully capture the developmental differences in UGT1A1 expression and metabolism observed in children [9],[10]. This review also did not attempt a quantitative meta-analysis. That choice was deliberate: the marked heterogeneity in study design, genotyping methods, and reported outcomes would have made pooled effect estimates unreliable. A narrative synthesis was therefore judged to be more appropriate for summarizing patterns and highlighting consistent themes in the literature [21]. Some practical challenges—such as variation in genotyping assays, differences in reporting quality, and difficulties in obtaining certain full texts—may have introduced a degree of selection or reporting bias [36].
Overall, these limitations point to the need for larger multi-center pediatric studies, more attention to rarer genetic variants, and the development of pediatric-specific clinical guidance to improve the translation of pharmacogenomic findings into practice. Addressing these gaps will be essential for moving from preliminary associations to clinically actionable, evidence-based recommendations that can safely guide irinotecan use in children.
UGT1A1 pharmacogenomics offers a powerful approach to individualizing irinotecan therapy in pediatric oncology, with strong evidence linking UGT1A1*28 and *6 polymorphisms to increased risks of severe neutropenia and diarrhea. Identifying these variants pre-emptively allows clinicians to adjust dosing strategies, minimizing toxicities while preserving efficacy. However, the dynamic developmental changes in UGT1A1 activity among children underscore the need for pediatric-specific guidelines and dosing algorithms. Progress in this field requires large, prospective pediatric trials, integration of polygenic risk models, and solutions to barriers such as cost, access, and clinician awareness. Future integration of UGT1A1 testing into pediatric oncology protocols has the potential to transform chemotherapy safety, reduce healthcare costs, and establish a foundation for broader pharmacogenomic implementation in children.