Oral Sphere
Journal of Dental and Health Sciences
Journal of Dental and Health Sciences
Received: 2025-01-06
Accepted: 2025-09-25
Published: 2025-10-01
Pages: 239-243
Background: Sturge-Weber syndrome is a congenital neurocutaneous disorder characterized by facial port-wine stain, seizures, intellectual disability, glaucoma, etc. After tuberous sclerosis and neurofibromatosis, it is the most prevalent neurocutaneous condition.
Case Presentation: A 16-year-old male patient presented with a unilateral port wine stain on the right side of the face and pain in the right side of the eye. On the clinical examination, there was a port-wine stain present extraorally and intraorally, and generalized bleeding on probing from the gingiva. The patient's past medical history revealed glaucoma for 6 years, and he is under medication. Sturge-Weber syndrome consists of ocular, neurological, cutaneous and oral manifestations. The most common manifestation seen on the unilateral side of the face and involves the intra-orally unilateral side. In the present case, there is a glaucoma in the right eye and a port-wine stain on the right side of the eye. Based on the Roach classification, our case fell under Type-I.
Conclusion: Sturge weber syndrome requires a multidisciplinary team work and all the treatment are symptomatic, there is no cure for the syndrome. Patient need ophthalmologist for the follow up due to risk of developing glaucoma and the dermatologist for the development of the lesion.
A facial cutaneous vascular nevus associated with leptomeningeal angiomatosis is the hallmark of Sturge Weber Syndrome (SWS), often referred to as encephelotrigeminal angiomatosis, a rare, nonhereditary illness [1].
First identified by SCHIRMER in 1860, the Sturge-Weber Syndrome was further defined by Sturge in 1878, who linked the neurological symptoms of the condition to changes in the skin and eyes [2].
Sturge-Weber syndrome is an uncommon developmental disorder that can result from somatic mosaic mutations in the GNAQ gene, which is located on the long arm of chromosome 9 [3].
The facial nevus is made up of many capillary-like vessels with thin walls. Angiomas, which are often limited to the pia mater and comprise numerous capillaries and tiny venous channels, are the neuropathological finding. The blood is transferred to the deep venous system via the larger medullary veins, which causes stasis and ischemic alterations, and there is a relative deficiency of superficial cortical veins [3].
Calcium deposits form in the cerebral cortex underneath the angioma as a result of changes in vascular dynamics brought on by the angioma. This may result in seizures, mental retardation, hemiplegia, or hemiparesis, the severity of which is determined by the degree of the damage [8]. It is pathognomic when the ophthalmic division's area is affected. Bupthalmos, hemianopia, horoidal hemangioma, and glaucoma can all be caused by ocular involvement [9].
For diagnosis, gadolinium contrast magnetic resonance imaging is the preferred neuroimaging method [4]. The incidence of SturgeWeber syndrome is estimated to be 1 in 20,000-50,000 live births. SWS affects males and females equally and there is no race predilection [3].
The importance of oral symptoms in SWS highlights the need for an oral health specialist to have a thorough awareness of this uncommon congenital condition. The known risk of intraoperative and postoperative hemorrhage may make routine dental and oral surgery more difficult [7].
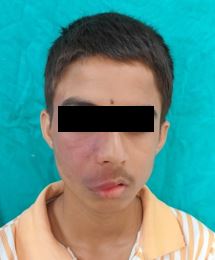
A 16-year-old male patient reported to the department with the chief complaint of facial asymmetry due to staining (Port wine stain on the right side of the face since childhood and generalized bleeding on probing for 3-4 months. The patient's past medical history revealed glaucoma for 6 years, and he is under medication. There was no history of mental retardation or convulsions, only a history of pressure or pain in the right eye. Extraoral examination showed a port wine stain on the right side of the face. It extended from the lower part of the forehead, eye, cheek, zygomatic arch, half of the nose, and philtrum and extended till the right side of the upper lip and the corner of the mouth Figure 1.



Not every patient with port wine stains on their face has Sturge- Weber angiomatosis. The development of the entire condition is only at risk for patients who have involvement along the distribution of the trigeminal nerve's ocular branch [5]. When both facial and CNS angiomas are involved then only it’s a SWS [6]. In 1992, Roach categorized SWS variants into three types: Type I: individual has a facial Port Wine Stain (PWS), leptomeningeal angioma, and may have glaucoma Type II: individual has a facial PWS, no leptomeningeal angioma, and glaucoma may be present, Type III: individual has leptomeningeal angiomatosis, no facial PWS, and glaucoma rarely present [7].
A "vascular steal phenomenon" may form around the brain's leptomeninges angioma, causing blood to pool and obstructing venous return, making it difficult for new blood to reach the affected areas of the brain that experience ischemic insult, atrophy, and calcifications. Recurrent seizures, status epilepticus, intractable seizures, and recurrent vascular events can all contribute to this theft. An increase in cortical ischemia can cause progressive calcification, gliosis, and atrophy, which can then cause seizures and neurological deterioration [4].
Among the differential diagnoses are Klippel Trenaumy-Weber syndrome, Von Hippel Lindau disease, Maffuci's syndrome, Rendu-Oslar-Weber syndrome, and angio- osteodystrophy syndrome [5].
In this case, there is glaucoma on the right side of the eye since 6 years and portwine stain on the right side of the face since childhood. There was no history of mental retardation and convulsation, only a history of pressure or pain in the right eye. Gonioscopy confirms the diagnosis of glaucoma in the right eye. Based on the Roach classification, our case fell under Type-I. According to a Sunil Timilsina et al. case report, a five-year-old girl has had a high-grade fever, headache, and widespread tonic-clonic convulsions for the past three days. According to the patient's mother, the infant was on antiepileptic medications (AEDs) when the first seizure happened, which happened at 4 months of age.
History also showed that the face had a reddish discoloration (portwine stain) from birth, which grew darker with time. The brain's right occipitoparietal area displayed mild T2 fluid- attenuated inversion recovery (FLAIR) hyperintensity on magnetic resonance imaging (MRI) [6],[7]. A brain computed tomography (CT) scan revealed bilateral white matter calcification, or calcified regions in the white matter on both sides of the brain, as well as a decrease in the amount of brain tissue (parenchymal volume loss) [8]-[10].
According to Azzeddine Laaraje's case report, an 11-year-old girl experienced mild left hemiparesis and drug-resistant epilepsy at the age of 18 months. Seizures remained poorly managed even after using several anticonvulsant drugs. Without facial port-wine stain or ophthalmological involvement, brain imaging showed classic findings of right cerebral hemihypotrophy, cortical calcifications, and leptomeningeal enhancement. SWS-III was diagnosed as a result of these unique radiological characteristics [11]. The Valbona Govori case study found that long-term seizure- free living is achieved when a patient with Sturge-Weber receives frequent treatment with 600 mg of carbamazepine. Seizures are controlled and consequences are avoided with effective early therapy [12].
Vigilant observation, steroids, radiation therapy, antimetabolites, injections of sclerosing solutions (sodium tetradecyl sulfate) into the gingival tissue, and surgical procedures such as LASERS (neodymium-doped yttrium aluminum garnet (Nd:YAG) laser, CO2 laser), cryosurgery (nitrous oxide gas at a temperature of -89°C at the probe tip), or electrosurgery are several methods used to treat gingival hyperplasia. In this instance, an extensive plaque control program (frequent deep scaling and root planing, encouraging patients to practice good oral hygiene, and using a mouthwash containing 0.2% chlorhexidine after brushing), along with a balanced diet regimen was followed to treat gingival hypertrophy [6].
Intracerebral vascular malformation results in neurological impairment [13]. Tram line calcifications, cortical atrophy, an enlarged ipsilateral choroid plexus, and pial angiomatosis are among the imaging findings [14]. While calcifications can be evaluated in depth on CT images, MRI is the best imaging modality. The development of seizures before the age of two years is associated with a bad prognosis in cases of refractory epilepsy and mental retardation [15], due to the larger involvement of brain malfunction.
Sturge-Weber syndrome treatment requires a multidisciplinary team effort for the patient to receive the right care and treatment. SWS manifests in a variety of clinical ways. The therapy of this ailment greatly depends on having a good understanding of this syndrome and the most recent treatment options available. To provide the patient with appropriate medical treatment, a multidisciplinary team works together. Caregivers should be given a thorough understanding of the disease as a whole. For patients to receive prompt and efficient treatment, caregivers must understand the repercussions of complications.
Despite the difficulties and the lack of understanding from others, living with Sturge-Weber Syndrome for the past few years has been an intriguing experience, unlike any other that most people have to face. Since the dawn of puberty, I have had a port wine stain that has expanded with the growth of my muscles. Since it is a part of my face, it located from my forehead and extending just below my lip line. Being port wine is more commonly known, it is hard to not feel different about that. To positive, it is a charming thing about my face, but when people are staring, it’s hard if I’m different about that.
Since my eyesight and glaucoma has been an issue since the past six years, I have had a hard time motivationally focusing on my work due to unmanageable pressure. I have also been on a constant stream of medications to try and ease the effects. I have just been really thinking about the future in a negative light if my eyesight will be unmanageable in the future. Another issue I have had with my is in my gums I have a lot of bleeding, which is a problem that has also made dental visits hard. I realize now that it is unmanageable to floss and I have to be more cautious with cleaning my mouth.
In SWS, or Sturge-Weber syndrome, I am required to see doctors from multiple specialties, including ophthalmology, dermatology, and dentistry. While SWS, or Sturge-Weber syndrome, cannot be cured, there is no need to lose hope. I trust my doctors to help me manage my symptoms for the best quality of life.